Spliceosome conserved sequences as seen through a G:U complement
Abstract
The spliceosome, comprised of small nuclear RNAs and proteins, is used by cells to splice out an intron in pre-mRNA and join its two flanking exons creating an mRNA. This installment of Snippets will be taking a closer, more detailed look at the conserved sequences of the Saccharomyces cerevisiae spliceosome as described by Madhani, H and Guthrie, C. (1992) and (1994). Of interest are two short complementary helical regions, where snU6 complements snU2 (helix 1or helix 1a +helix 1b)) and another where snU2 complements the conserved branchpoint of a pre-mRNA intron. The branchpoint is "used" by the spliceosome to form a lariat that in turn, is instrumental in splicing out the 5' end of the intron from the pre-mRNA.
Spliceosome
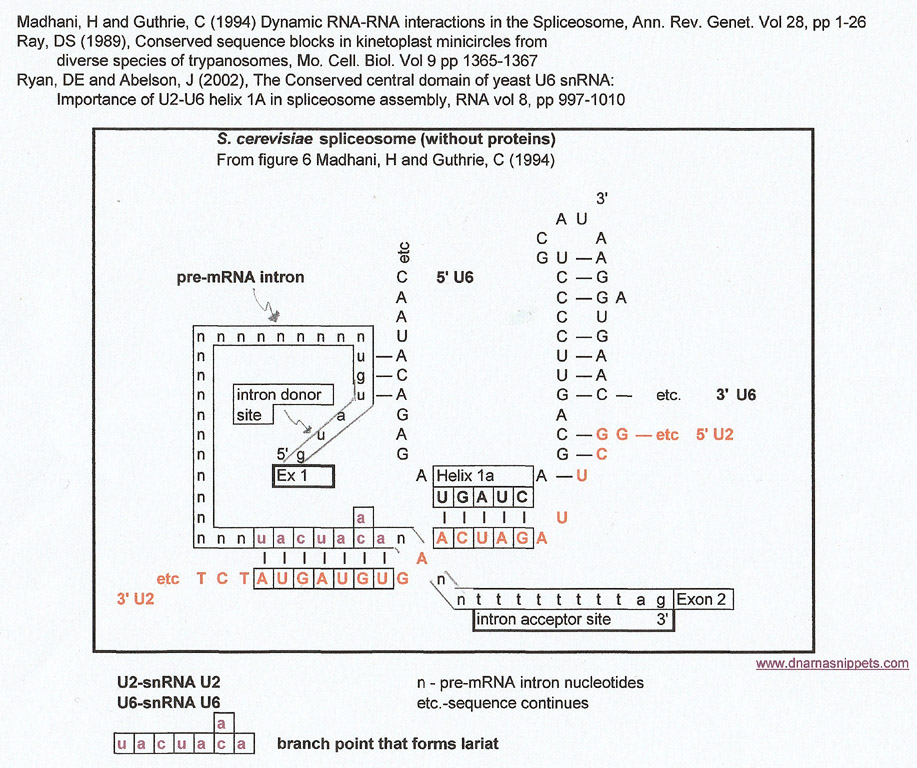
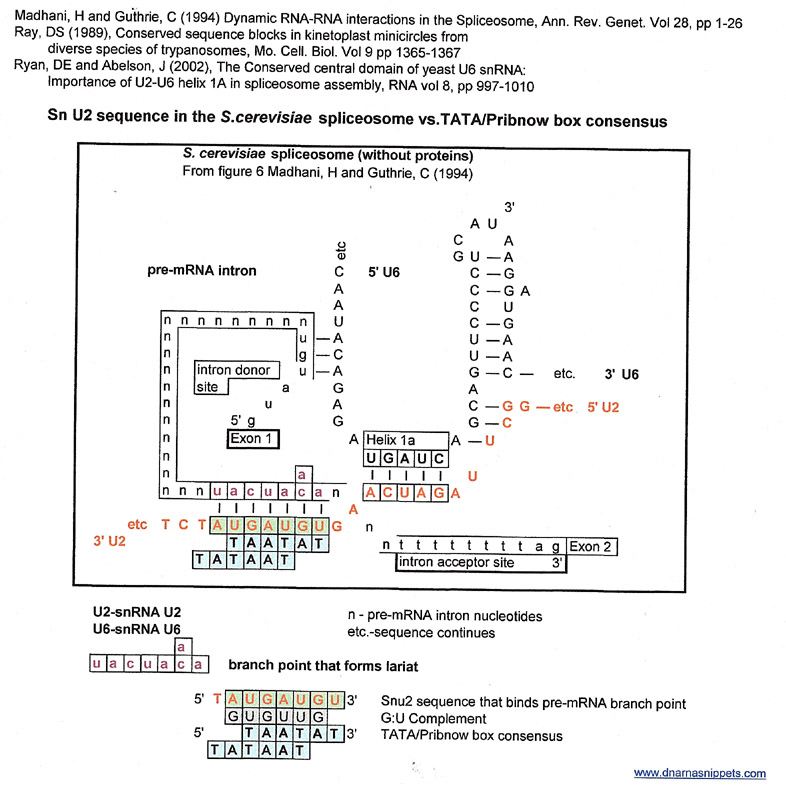
The graphic below is a stream-lined version of the model of the S. cerevisiae spliceosome from figure 6, (Madhani, H and Guthrie, C (1994)) without proteins or snU5 to better display the helical structures of snU6 with snU2 and snU2 with the branchpoint of a pre-mRNA. The pre-mRNA 5' intron donor site sequence is from Madhani's paper, and the 3' intron acceptor site sequence is from Lewin's Genes X page 579 (don't know if it is for S. cerevisiae pre-mRNAs).
This structure, with attendant proteins and other small nuclear RNAs is responsible for splicing out an intron at a conserved 5' donor site and a conserved 3' acceptor site in a pre-mRNA and ligating the two adjoining exons to form an mRNA. Pre-mRNAs may have more than one intron, but one is assuming that the spliceosome is "used" to cleave out each intron. A more detailed review of this mechanism can be found in any basic molecular biology text.
The graphic below will be used to make matches to other known conserved sequences using a G:U intermediate. For each set of resulting matches that will be made the same basic graphic will be used but with different highlighted sequences and different annotations. However, the yeast (S. cerevisiae) spliceosome RNA sequences (snU2 and snU6) will remain the same for each result
Methodology
The particular sequences of the Saccharomyces cerevisiae (yeast) spliceosome that will be used for comparisons will be shown in the above graphic and then, in another graphic, how, through a G:U complement a match is generated.
So, what is a match? In short a match to a sequence of interest is made by substituting a purine for a purine or a pyrimidine for a pyrimidine. To do this systematically, a G:U complement is made for each sequence of interest as described in the following section.
The section, "Generating a G:U complement" uses the sequence CCAGGG as an example of a sequence of interest..
Generating a G:U Complement to Make Matches
CCAGGG is a binding site of a retroviral primer and is not part of the spliceosome, but is being used to explain how a G:U complement is derived,
So, for some general "rules" that were used when making comparisons and searching for matches:
-All the nucleotide sequences used were considered as 'strings of beads" or letters in an "alphabet" that had no other meaning as to molecular chemistry, function or location, or species etc. The bases A,G,T,C were considered a 4 letter alphabet, and G,U a 2 letter alphabet.
-The G:U 2 letter alphabet is simply a tool to make matches and my use does not mean to imply that it is/was used in evolution, although it could have been.
-T and U were used interchangeably.
-If a match or matches were made to the sequence of interest, the temptation to say that "correlation was causation" was ignored and, the match sequence was not necessarily considered biologically related to the original sequence of interest.
-Sequences were analyzed both in the 5-'3' and 3'-5' direction.
-An uncommon nucleotide like di-hydroxyuridine (D) or pseudouridine (psi) was considered to be uridine because complementation was the only sequence interaction used in this kind of analysis.
- Proteins were not considered in any of the methods.
-Using this method, C and A are considered complementary.
-Making match results are only as good as the sequences used. If these are not accurate or real, this author has no way of knowing so with that understood, the results will be presented.
As an example, to create a G:U intermediate of CCAGGG, a complement of G's and Us (gray background) ( 2 letter alphabet) was generated. In the next step, reverse complements (4 letter alphabet) were generated from the G:U complement. These were the "matches". Below, one can see that some matches to CCAGGG were telomere repeats for humans and Tetrahymena . This does not imply that there is a biological or causal relationship between the retroviral binding site (CCAGGG) and the telomere repeats TTAGGG, TTGGGG). (However, see "Snippets 16 Telomere Repeats through a G:U Intermediate")
Generating a G:U Complement
| Anti-pas sequence of Retroviral primer | C | C | A | G | G | G |
| G:U complement | G | G | U | U | U | U |
| Reverse complement | C or T or U | C or T or U | A or G | A or G | A or G | A or G |
Using this methodology, some of the following "matches" for CCAGGG could be made
| Anti-pas sequence of Retroviral primer | C | C | A | G | G | G |
| G:U complement | G | G | U | U | U | U |
| homology | C | C | A | G | G | G |
| match | T | C | A | G | G | G |
| Telomere rpt humans, mice | T | T | A | G | G | G |
| match | C | C | G | G | G | G |
| match | C | T | A | G | G | G |
| match | C | T | G | G | G | G |
| Telomere rpt. Tetrahymena | T | T | G | G | G | G |
| match to G:U complement | G | A | T | T | T | T |
| match to G:U complement | A | A | C | C | T | C |
| match to both | T | T | T | T | T | T |
To reiterate, the fact that a match could be found between the retroviral primer anti-pas site and the human and tetrahymena telomere repeat did and does not mean that there is any known biological or causal relationship between the two.
It is important to note that a match can be made to both the positive strand (the sequence of interest) and its G:U complement (see last line of above chart of results). This author made the arbitrary decision that the split sections of a match made between the both the sequence of interest and its G:U complement (match to both) had to be at least 4 or 5 nucleotides in length.
In essence, one is substituting a purine for another purine or a pyrimidine for another pyrimidine to make a match. One could use the letters "Y" for pyrimidine or "R" for purine instead of G and U but this author liked G and U. Even the numbers 1 or 0 could be used provided they were each defined as a specific set of nucleotide bases (ie 1 stands for A or G and 0 stands for C, U, or T).
Briefly put, not only can one substitute a purine for another purine, or a pyrimidine for a pyrimide, but one has to accept that C and A are complementary using this method.
Unlike homologies, there can be more than one match for any given sequence of interest. This is important.
One author, working with homologies used an 80% similarity as a criteria, and this author has kept that at the back of her mind when subjectively making matches.
This author used a number of papers from the 1980s because she found that using large genomic databases had the following problem. She would locate a sequence in the large database and then when she went back to find it, the database had been updated and the sequence was not at the same location. (The nucleotide numbers were different). So, if the database had a manuscript reference for the original sequence and the original sequence was still the same as the one in the database she used the original paper as a reference
The complete sequence of Trypanosoma equiperdum clone 818mB minicircle, (GenBank EU155059.1) was used for CSB-3 and kinetoplastid gap comparisons. Other T. equiperdum clones had the same sequences around the CSB-3 region, but their base numbers were different, so the 818mB minicircle clone was used for all comparisons in this installment of Snippets.
"Snippets" should not be confused with Single Nucleotide Polymorphisms (SNPs). I was simply trying to convey that I was looking at short strings of nucleotides.
"Aha!' you say, This methodology has a problem. If one substituted a purine for a purine and a pyrimidine for a pyrimidine, a choice of only two or four letters, of course one would find matches on a random basis."
Maybe, maybe not, given enough letters, but that is why I picked conserved sequences to study. As I explained in the discussion of Snippets 15a, DNA and RNA sequences obey the same dictates of evolution as any organism's continuation. In other words, those sequences that are necessary for replication such as origins of replication, or primer sites are necessary for the continuation of an organism on a genetic level. There is a tendency to look at the string of letters or bases in non-coding DNA or RNA as an amalgam of random events and to treat them statistically as such. But, in terms of evolution or the continuation of a replicating species this couldn't work.
Results
Before continuing, I'd like to explain, in more detail, the importance of the Conserved Sequence Bloc 3 (CSB-3) and its overlapping kinetoplastid gap, a single stranded DNA region that is probably an RNA primer site in the mitochondria of kinetoplast minicircles. (Ntambi,P et al (1986))
Why elaborate on such a rather obscure sequence? It's simple. When I started searching for short conserved sequences (hence the name "Snippets") I entered "conserved sequences" into the ncbi/nlm search engine and a reference describing CSB-3 in Trypanosomes appeared on my screen. Using that initial sequence, I continued my observations.
In the mitochondrion of of each kinetoplast, there is a network of thousands of DNA minicircles and each minicircle has a universally strictly conserved CSB-3 sequence that is considered an origin of replication CSB-3 is thought to be one of two origins of replication because it is one of two binding sites for the Universal Minicircle Binding Protein a sequence specific binding protein involved in the replication of the kinetoplast C. fasciculata (Abu Eineel, et al (1999)). Whether this is also true for T. equiperdum has not been demonstrated, but since CSB-3 is universally conserved in all kinetoplast DNA minicircles, it seems probable that it is an origin of replication for T. equiperdum also.
Unlike mammallian mitochondria, the one mitochondrion in kinetoplasts replicates when the kinetoplast replicates and the many minicircles are part of a catenated network, quite different from bacteria and archea.
One can guess that the mitochondrion is the remains of a symbiont, but the question is, which symbiont had such a replication process? Without going into detail about how the DNA minicircles replicate, this part of the Snippets installment will concentrate on the CSB-3 sequence: (GGGGTTGGTGTAAT).
To further complicate matters, Ntambi, P and Englund (1985) showed that embedded in CSB-3 for each newly replicated T. equiperdum minicircle was a single-stranded region (TTGGTGTAAT) that is the complement of the kinetoplastid gap. They deduced the gap was an RNA primer site (Ntambi, P et al. (1986)). .In the following results, I use TTGGTGTAAT as the sequence for the kinetoplastid gap since their paper did not show which RNA nucleotides were used as primers to fill the gap and it doesn't really matter using the G:U complement methodology.
from figure 6 Ntambi, P et al (1986)
| T | G | T | Kinetoplastid Gap | C | C | C | C | T | T | T | |||||||||
| A | C | A | T | A | A | T | G | T | G | G | T | T | G | G | G | G | A | A | A |
| CSB-3 | |||||||||||||||||||
Spliceosome (S. cerevisiae) vs kinetoplastid Conserved Sequence Bloc 3 and the kinetoplastid gap
through a G:U complement
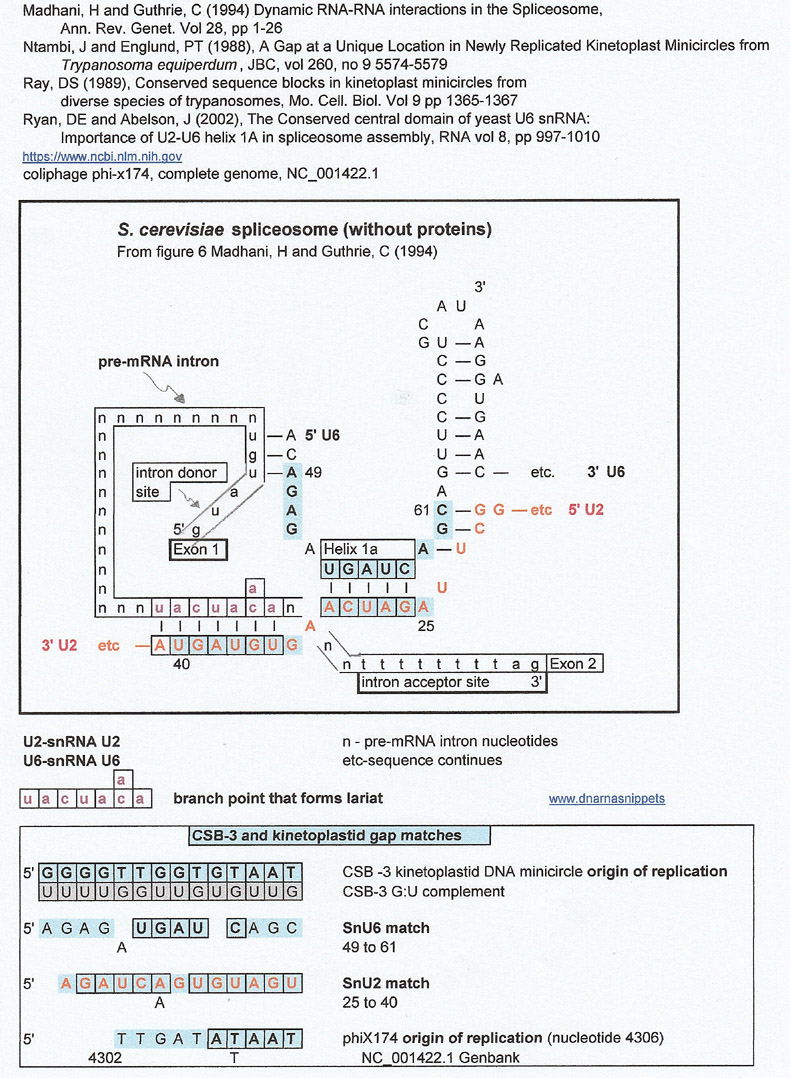
I am showing this result because the region around helix 1a where SnU6 and SnU2 are complementary are a match for the conserved origin of replication for kinetoplast mitochondrial minicircles.
The fact that the kinetoplastid gap is a match for the origin of replication (ori) of phiX174 a single stranded DNA phage (with a DNA double stranded replicative form) is intriguing.
This author tried to find oris of other ssDNA phages but could not retrieve the sequences from on-line databases. However, in going through the literature, the author found an article by Siebenlist, U (1979) that showed a good match between the helix 1 region to the T7 A-1 regulatory promoter where transcription of the T7 coliphage starts. Since the article was from 1979, she confirmed the transcription start site with the ncbi database (which probably used the 1979 data). See below. This prompted a closer look at E. coli and B. subtilis promoters and eventually chloroplast promoters..
So, in summary, the conserved kinetoplastid gap, an ori for a T. equiperdum DNA minicircle, phiX174's ori and the transcription start site for T7 coliphage (see result below) are all matches to one another and to the helix1a and helix 1b (58-61 of SnU6) portion of the spliceosome.
E. coli oris were not a match.
Since the T7 A-1 transcription site showed a match to its promoter and the kinetoplastid gap looked like a match to the conserved promoter TATA/Pribnow box a further set of comparisons were made to E. coli and B subtilis promoters.
(I am going to front load the results with matches to full promoter sequences and then show matches for the conserved TATA box/Pribnow box and "-35 elements".
Spliceosome (S. cerevisiae) versus Promoters
Promoters, the binding sites for RNA polymerases are essentially regulatory regions for transcription of pre-mRNAs. The pre-mRNAs undergo a process, using the spliceosome, in which their introns are excised out and the bordering exons are joined to make a final mRNA. Prokaryotic promoters have two conserved sequences, the Pribnow or TATA box at approximately 15 nucleotides 5' to the transcription start site (-15), and the "-35 element", 35 nucleotides 5' to the transcription start site (-35). +1 is considered the transcription start site. Although, since the 1970s and 1980s, other transcription factor binding sites have been demonstrated, this installment of Snippets will confine itself to the TATA box and what was termed in the 1980s the "-35 element".
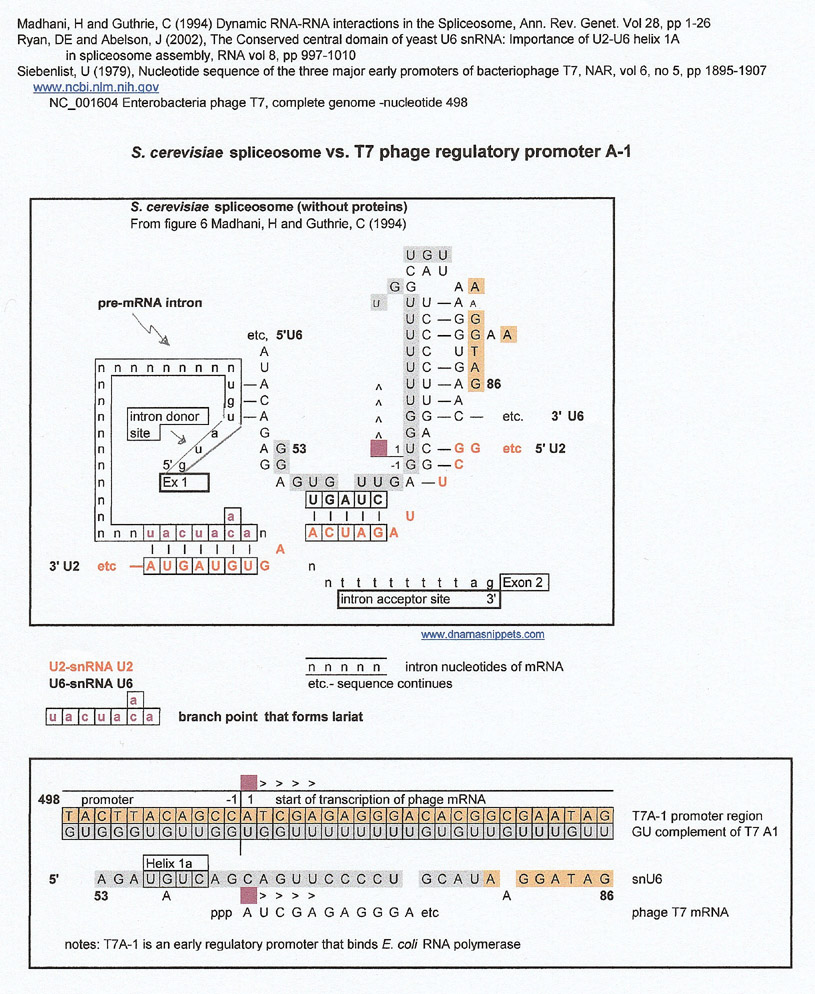
I decided to look for matches between the spliceosome sequences and promoters and was surprised to find a match for the T7A-1 phage regulatory promoter, The match is confusing at first because it is between the G:U complement (gray letters) of the phage promoter/ transcription start site and Sn U6.
S. cerevisiae spliceosome vs. T7 phage regulatory promoter A-1
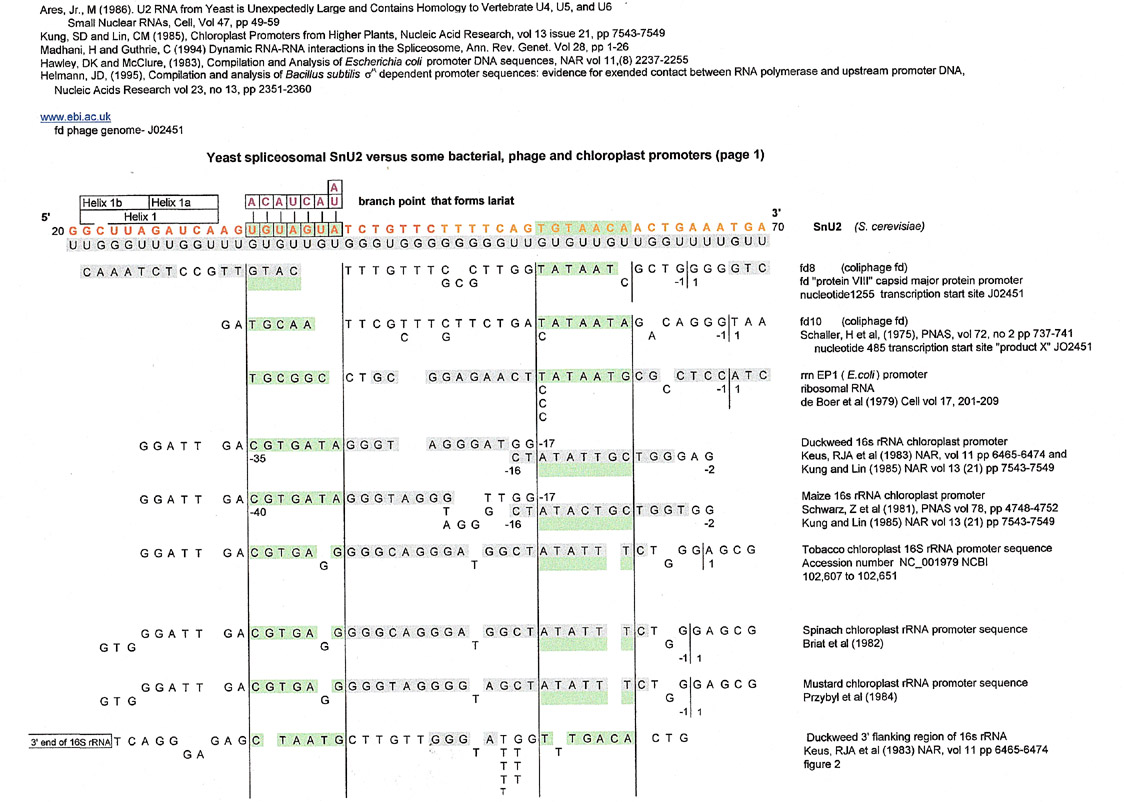
It didn't take long for this author to decide to look at other promoters and realized that she could find some interesting matches to snU2, not snU6. The matches were made to the snU2 sequence that was complementary to the lariat and helix1a.part of the spliceosome (nos 20 to 70)(Ares, Jr., M (1986))
Spoiler Alert: Below are the results for this region; (the snU2 sequence is in orange letters). There was a varied assortment of matches, but the most numerous were for higher plant chloroplast 16s ribosomal RNA promoters. Of course, there are probably many more, but this author couldn't get access to her usual e-journal databases due to the pandemic.
Chloroplasts are thought to be symbionts of photosynthetic cyanobacteria and once again, this author couldn't get access to sequence data to look at their rRNA promoters. It might also be worth investigating bacterial operons.
Please note that in the graphic above, SnU2 is 3' to 5' and in the graphic below, is 5' to 3'. Letters in gray are derived from the GU complement directly.
Individual sequence references were found in the Hawley, DK and McClure (1983) , Kung, and Lin (1985) and Helmnn, JD (1985) compilations and verified whenever possible.

This author is going to include the following list of results of matches to bacterial and phage promoter TATA boxes and CSB-3 sequences even though the matches are shorter in length than the above promoter results,.because CSB-3 is considered a conserved origin of replication. Of note is that many of the matches are in the 3'-5' direction and that the matches for conserved sequences (green and blue) are to the plus strand of snU2.
S. cerevisiae spliceosome vs. bacterial and phage promoter TATA boxes
page1crop40percent.jpg)
page2crop40percent.jpg)
As one can see there were some matches from some promoters for B subtilis and E. coli to CSB-3 and the kinetoplastid gap. The most prevalent match was to the smaller kinetoplastid gap, the single stranded RNA primer site in T. equiperdum minicircles that is embedded in CSB-3, but some of the matches extended to CSB-3.
After a brief, incomplete survey, it seemed that matches to phage promoters, operon promoters (lac1, trp tna) and ribosomal RNA transcription start sites were better than to other E. coli and B subtilis promoters.
But these matches were only from a few of the many promoters compiled in the Hawley and Hellman papers.
E. coli ribosomal RNA promoters could be matched in both the 5'-3' as well as 3'-5' directions, but did not show a definite correlation with bacterial or phage ribosomal RNA or transcription start sites of tRNAs as there wasn't enough data. It was hard to find promoters for defined operons (regulatory regions) in the databases, but it would be useful to pursue this angle as some tRNAs sequences are found together under the regulation of one operon. Also lacl, the first gene in the lac operon, and some of the rRNAs showed a small overlap in their 5'-3' matches at the -6, -7, -8, -9 nucleotide locations. This overlap in a match has been found in other comparisons (see RSS comparisons below).
As shown above, the transcription start site of T7 coliphage (T7A-1) is a good match for SnU6, and T7A-3, another early T7 regulatory sequence, is a match for the SnU2 sequence.
The coliphage T7 early regulatory promoters bind to E. coli RNA polymerase, probably as part of their "takeover" of the E. coli metabolic machinery.
The sequence reported for Tn5-neo was an excellent match, but after skimming the original article some questions were raised about the actual sequence reported as a promoter. The authors, Rothstein, SJ and Reznikoff, WS (1981) were closely examining the 5' and 3' repeats of transposon5 to show that they were same except for one nucleotide. They decided that a certain region of these almost homologous repeats was a promoter because it looked as though it contained a probable TATA and "-35 element" box. Whether their decision was correct or not, the above result is that the terminal repeats of Tn5 are a good match for comparison with SnU2. It would be useful to look at other transposon inverted repeats.
Which raises the never-ending question, "how many letters make a good match?" Since one can't quantify these results other than the subjective decision to accept 80% of the nucleotides as a match, it is hard to justify these results in the scientific arena. And, if a match indicates something real, then what does it mean?
Most of the reference papers used in this installment of Snippets were from the 1970s and 1980s and at that time databases were much sparser than now, so some of the transcription products were not specifically identified. Sequence deletions or mutants were used to show that the protein product was deleted and a pathway obstructed, but the actual protein remained to be identified. However, the nucleotide sequences themselves, derived using the Sanger method, or nucleases (taking weeks), seem to still be good.
Most of the promoters were not matches, so, once again this highlights the scientific weakness of making matches.. In other words, were there enough nucleotides involved so that matches could be randomly made? And, helix 1a and the branchpoint are only five nucleotides increasing the chances of a random match. But since these five nucleotides are conserved and are a protein binding site this author continued her comparisons. It is noteworthy that the branchpoint is used to form a lariat, the result of a splice at the 5' end of the pre-mRNA intron , and as shown below is a complementary match for the conserved RSS heptamer, also a splice site.
It would be useful to pursue the transcription promoters of other non-coding RNAs, like tRNAs, rRNAs snRNAs or other RNA polymerase binding sites from prokaryotes. Since, according to Lewin's Genes X (2011), only prokaryotes have operons where one regulatory region might affect a series of genes (tRNAs, genes affecting one metabolic pathway) it raises the question of what are their origins?
Eukaryote regulatory regions occur uniquely for each gene. (Lewin's Genes X (2011)) but this was not pursued. by this author.
After looking for matches to the TATA box conserved region this author compared SnU6, not SnU2 to the other conserved sequence in promoters, the "-35 element" : TTGACA.
Again, a surprise.
Spliceosome (S. cerevisiae) versus the conserved -35 element of Bacterial and Phage Promoters
crop40percent.jpg)
It turns out, quite unexpectedly that there were two matches in tandem, or cis orientation, for the conserved TTGACA sequence going from SnU6 nucleotides 53 to 65 that encompass helices 1a and 1b (helix 1). Keeping that in mind, comparisons for both sequences were made when looking at promoters of E. coli and B. subtilis and their phages. Most of the matches for E. coli were to phages in the promoter 3'-5' direction. Matches for B. subtilis seemed to involve tRNA promoters and two matches were involved in the sporulation process.
(Author's note: just to clarify some of the results, "phi X a" is the promoter for phi X's protein A, the major capsid protein, and phiX D is the promoter for "product D", that was not identified in the paper. Unfortunately, the papers are old and this author did a literature search for phiX D to see if it had been identified, but couldn't find anything.)
The bases/letters in gray are matches made to the G:U complement itself, and, as can be seen for phage 434PR, there is a match made using an overlap of two bases.
crop40percent.jpg)
Interestingly, the B. subtilis promoters for glutamine synthetase, the tryptophan operon and the trnS (tRNA) operon (that contains seven tRNAs) had matches for both the TATA and "-35 element" conserved sequences.. A more thorough set of comparisons needs to be done.
What are the odds of the following?
In parting from this discussion of conserved sequences of promoters I leave you with the following. If you take the conserved "-35 element" and the conserved TATA box sequence and delete the variable promoter sequences in between, you get TTGACATAATAT, which through a G:U complement is TTGGTGTAATAC, the conserved kinetoplastid gap complement and 2 flanking nucleotides of T. equiperdum. Of course, this brings up the problem of looking at short sequences. Is this a random find or does it indicate something because the sequences involved are conserved? And, why, if the -35 element and TATA box sequences are somehow historically related to the sequence of the kinetoplastid gap, did the gap sequence get split and eventually used as an RNA polymerase binding site(s).?
On to the next observations.
Spliceosome (S. cerevisiae) versus T cell receptor and immunoglobulin recombination sequence signals
As described more thoroughly in Snippets 20, T cell receptors and immunoglobulins are comprised of heavy chains, and light chains (plus a constant region that won't be discussed further). Light chains have two sequences; a V (variable) gene sequence and a J (joining) gene, and heavy chains are comprised of V gene, D (diversity) gene and J gene sequences. Each V gene for instance is selected from an array of V genes and each J gene is selected from an array of J genes, and likewise for the D gene. They are then recombined to eventually form part of the final T cell receptor and or immunoglobulin sequence.
Having a V family, D family and J family of genes allows for a diverse selection of T cell receptors and immunoglobulins. For heavy chains, D and J are joined first, followed by a V+DJ recombination.
To facilitate recombination, each V, J and D gene has a conserved recombination signal sequence (RSS) that acts by complementation (and proteins) to align the gene so it can be recombined and ligated to the other genes to form the final product. Each recombination signal sequence (RSS) has a conserved heptamer and nonamer. The V gene is followed by the conserved (consensus) heptamer CACAGTG plus 23 nucleotides and then a nonamer of ACAAAAACC. A D gene has both a 5' heptamer and 3' heptamer on either side of it and the J gene is preceded by a 5' heptamer. Graphics are below
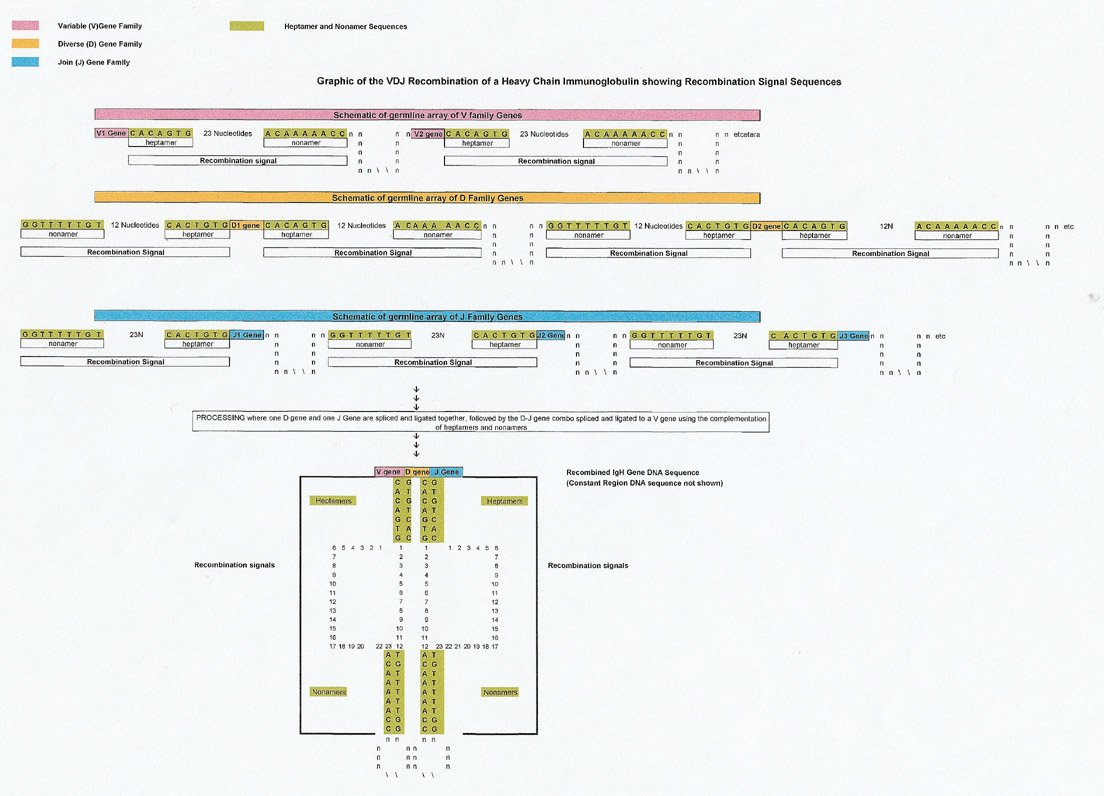
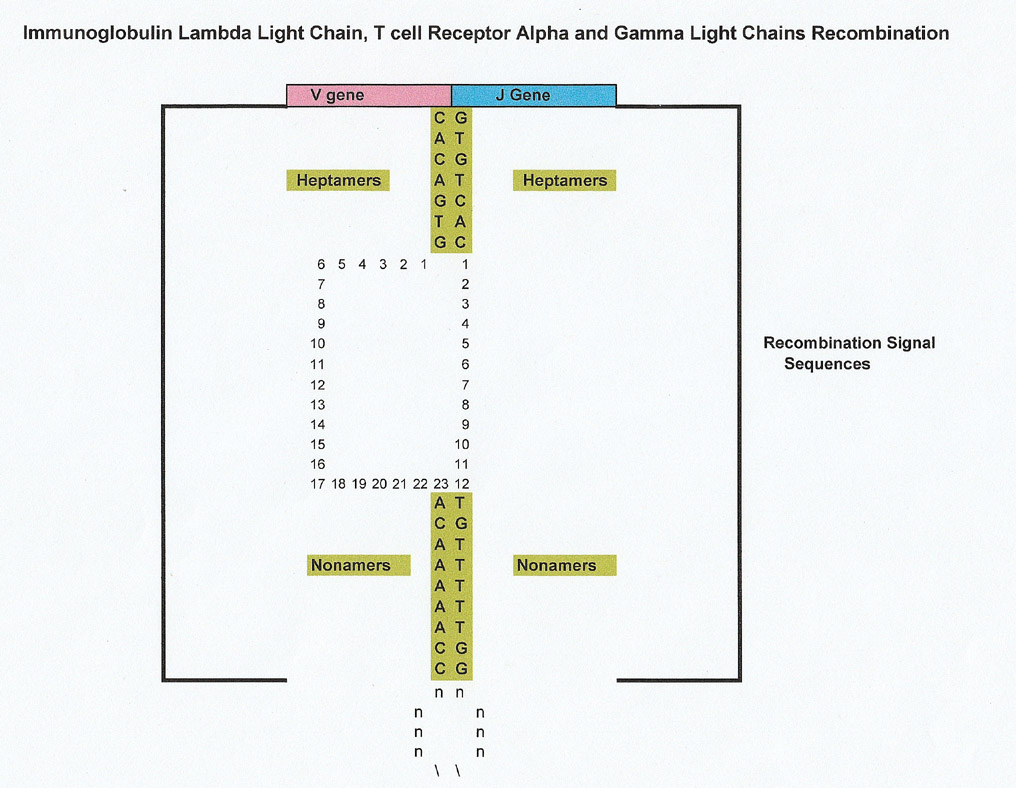
Spliceosome (S. cerevisiae) versus Recombination Signal Sequence (RSS) Conserved Heptamers and Nonamers

For the comparison below, the SnU2 sequence was extended in the 5' and 3' directions (sequence from Ares, Jr., M (1986)). Then a very incomplete cursory survey of the various T cell receptor and light and heavy chain loci was made looking for matches and some were found. The branchpoint and its SnU2 complement seemed to be a good match for the heptamer preceeding some D and J genes which is intriguing because the branchpoint is part of the lariat formation that is the result of the cleavage of the 5' intron site of the pre-mRNA. Don't forget that the heptamer is also a cleavage site.
At first glance, the results show that a complement of an SnU2 region 3' to the lariat is a match for some D genes from T cell receptor beta, T cell receptor delta and IgH. The 5' heptamer next to the D genes seem to be a match derived from the G:U complement directly (gray letters), and the 3' heptamer seems to be a match to the sequence of interest.
But, the results below are troublesome for the following reasons
There is no control random sequence to show that these results weren't random.
Many of the database V, D and J genes are predicted,
Loh's paper (1988), was useful but his TRDV-2 gene differed from the database. Was that because he probably deduced V sequences from various transformed thymocyte cell lines that were similar to germline TRD V genes or was the ncbi database incorrect? It's hard to choose sometimes what sequence to use. (The author used the germline sequence from Loh's paper),
It is interesting that the V gene sequence matches from T cell receptors were next to heptamers that coincided with the 3' heptamers of D genes. No conclusions can be drawn because all the T cell receptor and immunoglobulin databases were not searched thoroughly and the results are "spotty".
Of interest is that all the complete D gene matches for T cell receptors start just 3' to the branchpoint and that the D gene for IgH has overlaps in that region. The overlaps were reminiscent of kinetoplast gRNA overlaps which could be shown to be matches to some RSSs.. (see Snippets 20)
The SnU2 (S. cerevesiae) sequence versus the Recombination Signal Sequences of T cell Receptor and Immunoglobulin Genes
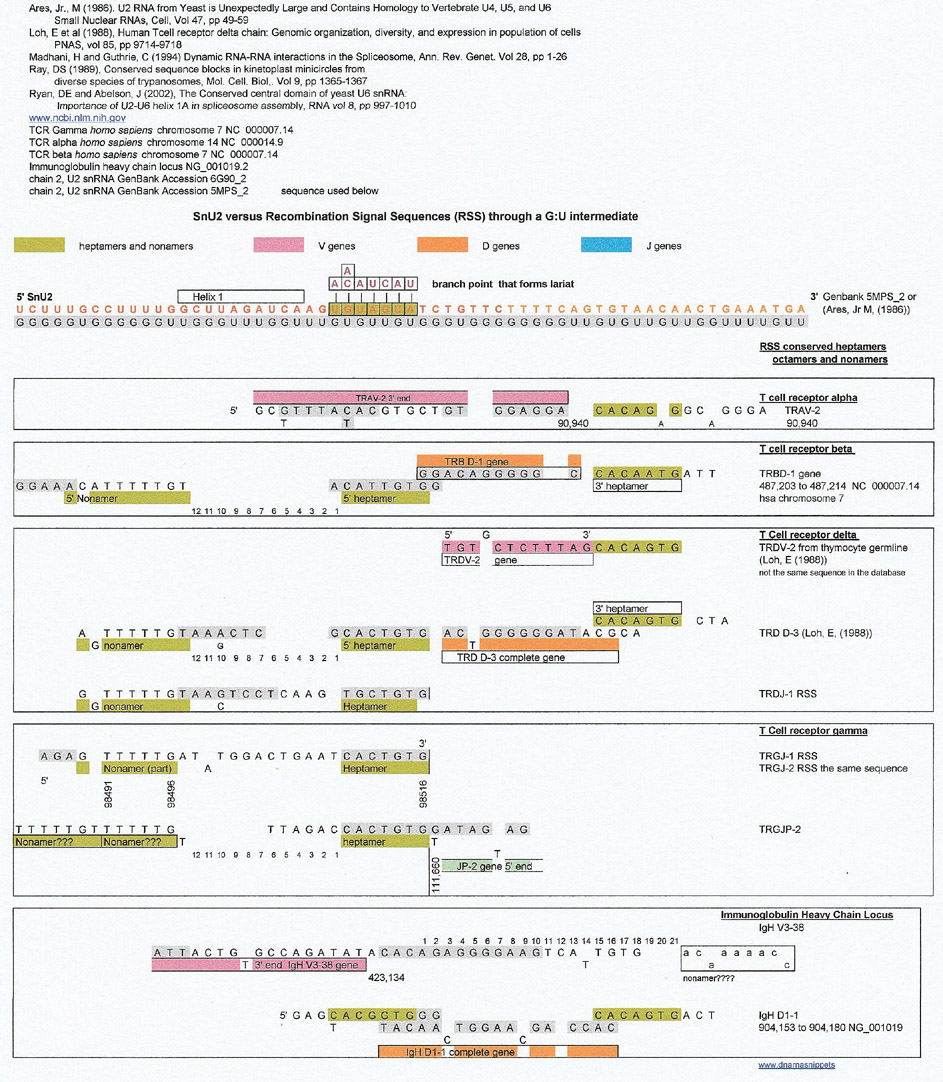
So, since some of the RSS matches involved overlapping sequences (IgH D1-1) this author, using a very bad scientific procedure; reasoning by analogy, decided to look at kinetoplast guide RNAs that create long RNA templates by overlapping sequences. If you go back to Snippets 20 you can see that there are a number of matches of overlapping gRNA sequences to the RSS sequences of T cell receptors and IgH sequences. Please remember that the overlapping regions are not complementary, but are duplicates of purines or pyrimidines.
The SnU2 (S. cerevisiae) sequence versus some kinetoplast guide RNA (gRNA) overlap regions of Leishmania tarentolae
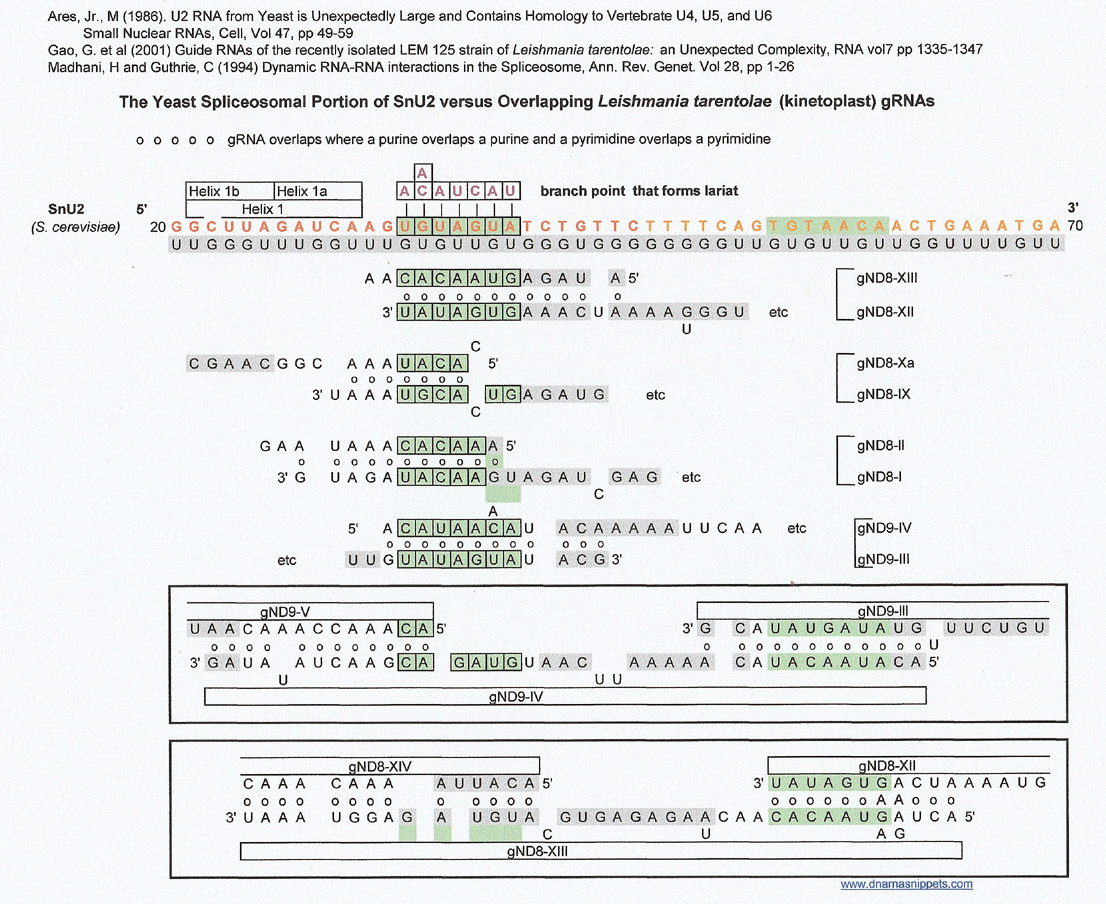
The overlaps were constructed by Gao, et al, and I am not sure what conclusions they drew from their results.
Since I was looking at the sequences used for splicing out introns, I decided to look at archael pre-tRNA introns which were easily available in Marck, C and Grosjean, H.'s 2003 paper.
Spliceosome (S. cerevesiae) SnU2 versus archael tRNA introns
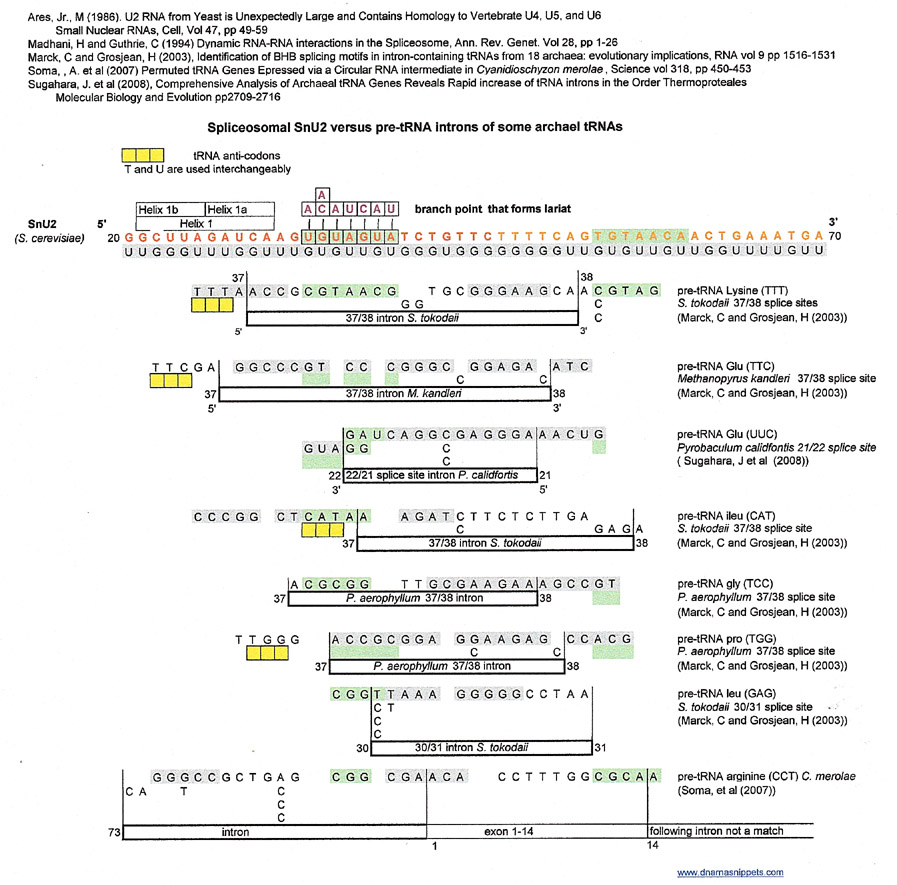
Most matches for these archael introns seem to be from the G:U complementary strand and occur at the 37/38 splice site of each pre-tRNA and include a match to the branchpoint that forms a lariat region of snU2. The branch point/ lariat, conserved in yeast is used by the spliceosome to cleave the 5' end of the pre-mRNA intron away from its neighboring 5' exon.
The example of pre-tRNA arginine (CCT) is interesting because Soma et. al. posit that some pre-tRNAs of C. merolae are circular and that in this example, the intron between the last (73) and first (1) nucleotide in the circular pre-tRNA is cleaved out to form the final tRNA for arginine.
Discussion
If one accepts that there can be more than one match to a sequence of interest, in this case the snU2 and snU6 components of the the S. cerevisiae spliceosome, then one has to contemplate what that means. The question of why one can find matches to the disparate sequences of promoters, D genes, archael pre-tRNA introns cannot be solved for now. Are the results really good luck random finds, or do they actually indicate something of the evolution of a given sequence?
That is why this author has chosen to look at conserved DNA and RNA sequences, preferably known origins of replication (ori) and RNA primer sites which must be conserved if the DNA or RNA of an organism is to replicate. I have deliberately ignored proteins in Snippets to make life simpler when making matches.
Why use a G:U complement to make matches? It is not really necessary to do so, as one can simply substitute a purine for a purine or a pyrimidine for a pyrimidine and assume C and A are complementary. It was easier for this author to get a handle on things once she realized that she was going to treat the bases as beads that could complement one another and that U complemented with A or G. and that U and T were interchangeable. This brings up the admonishments that "T doesn't complement with G" and "how did a U become a T in your sequences?" That is why, simply ignoring, for the present, all biological mechanisms and treating the sequences as strings of beads, using G and U as complements seemed the way to continue. In essence, a four letter alphabet was made into a 2 letter alphabet and then back out into a four letter alphabet to make matches. Did the early evolution of sequences use a G:U intermediate? Who knows. For this author it is simply a tool to find matches.
Although, if you look at sequences of certain tRNA primers in reverse transcription, or fMet (CAU) of tobacco chloroplast (Ohme, M et al (1984)) you can see that both the A-U complement as well as the G-U complement are used in the formation of the molecule's cloverleaf structure. There are probably many more examples of this in RNA molecules. I bring this up, just to show that the U-A and U-G complements can be used in the same molecule and if this molecule is somehow transcribed to DNA the A will complement T and the G will then complement C.
The results bring to mind the old parable of the five blind men and the elephant. The first man feels the elephant's foot and decides it is a tree trunk, the second feels the ear and thinks it is a sail, the third feels the trunk and decides it is a hose the fourth feels the tusk and concludes it is a hard stick and the fifth feels the tail and thinks of a rope. Of course, because they cannot see they cannot comprehend it is a whole big elephant and they have been only feeling the parts. In this case, if one accepts the above results and those in other installments of Snippets, the whole picture is impossible to see because DNA and RNA sequences are billions of years old and have passed through every species or organism that has propagated and populated our planet.
References
Abu-Eineel et al, (1999), Universal Minicircle Sequence-Binding Protein, a Sequence-specific DNA-binding Protein That Recognizes the Two Replication Origins of the Kinetoplastid DNA Minicircle, The Journal of Biological Chemistry, vol 274, no 19, pp 13419-13426.
Ares, Jr. M (1986), U2 RNA from Yeast is Unexpectedly Large and Contains Homology to Vertebrate U4, U5, and U6 Small Nuclear RNAs, Cell, vol 47, pp 49-59.
Gao, G. et al. (2001), Guide RNAs of the recently isolated LEM 125 strain of Leishmania tarantolae, an unexpected complexity, RNA vol 17, pp 1335 -1347.
Hawley, D and McClure, W (1983), Compilation and analysis of Escherichia coli promoter DNA sequences, Nucleic Acids Research, vol 11, No 8, pp 2237-2255.
Helmann, JD, (1995), Compilation and analysis of Bacillus subtilis sA-dependent promoter sequences: evidence for extended contact between RNA polymerase and upstream promoter DNA, Nucleic Acids Research, vol 23 no 13, pp 2351-2360.
Lewin's Genes X, Krebs JE, Goldstein, ES and Kilpatrick, S, eds., (2011), 10th edition, Jones and Bartlett, publishers
Loh, E et al, (1988), Human T cell receptor delta chain: Genomic organization, diversity and expression in population of cells, PNAS, vol 85, pp 9714-9718.
Madhani, H and Guthrie, C, (1992), A Novel Base-Pairing Interaction between U2 and U6 snRNAs suggests a Mechanism for the Catalytic Activation of the Spliceosome, Cell vol 71, pp 803-817.
Madhani, H and Guthrie, C, (1994), Dynamic RNA-RNA interactions in the Spliceosome, Ann Rev. Genetics, vol 28, pp 1-26.
Ntambi, J and Englund, P (1985) A Gap at a Unique Location in Newly Replicated Kinetoplast DNA Minicircles from Trypanosoma equiperdum, The Journal of Biological Chemistry, vol 260, no 9, pp 5574-5579.
Ntambi, J et al (1986), Ribonucleotides Associated with a Gap in Newly Replicated Kinetoplast DNA Minicircles from Trypanosoma equiperdeum, the Journal of Biological Chemistry vol 261, pp 11890-11895
Ohme, M. et. al., (1984), Locations and sequences of tobacco chloroplast genes for tRNAPro(UGG), tRNATrp(CCA), tRNAfMet(CAU) and tRNAGly(CCC),...Nucleic Acids Research, vol 12, no 17 pp 6741-6749.
Qin, D et al (2016), Sequencing of lariat termini in S. cerevisiae reveals 5' splice sites, branch points, and novel splicing events, RNA vol 22 pp 237-253.
Ray, DS (1989), Conserved sequence blocks in kinetoplast minicircles from diverse species of trypanosomes, Molecular Cell Biology vol 9 no 3, pp 1365-1367.
Ryan, DE and Abelson J. (2002), The Conserved central domain of yeast U6 sn RNA: Importance of the U2-U6 helix 1A in spliceosomal assembly, RNA, vol 8, pp 997-1010.
fd phage genome: J02451
https://www.ncbi.nlm.nih.gov sequences
Chain 2, U2 snRNA GenBank PDB: 5MPS_2
Coliphage phi-X174, complete genome GenBank NC_001422.1
Enterobacteria phage T7, complete Genome Genbank NC_001604.1
Immunoglobulin heavy chain locus GenBank NG_001019.2
T cell receptor beta (TRB), homo sapiens chromosome 7 GenBank NC_000007.14
T cell receptor delta (TRD), homo sapiens chromosome 14 GenBank NC_000014.9
T cell receptor gamma (TRG), homo sapiens chromosome 7 GenBank NC_000007.14
Trypanosoma equiperdum clone 818mB minicircle, complete sequence, GenBank EU155059.1